
The Materials-by-Design approach relies on strong theoretical capabilities to predict the properties of novel materials. Unfortunately, traditional ab initio techniques for calculating the electronic structure of materials are powerless when the lattice mismatch between two crystals leads to the absence of periodicity, as observed between many of the interface quantum materials that are the focus of PARADIM’s in-house research.
|
|
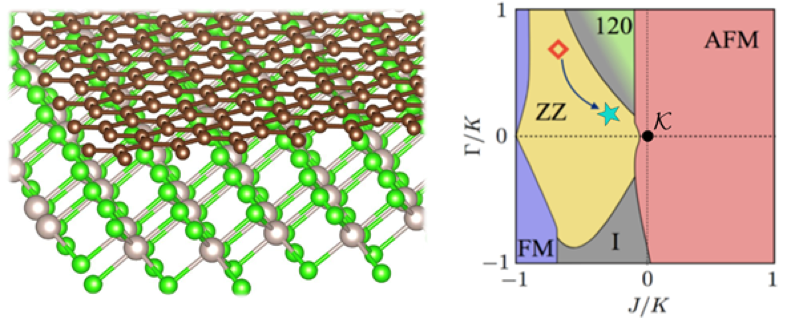
Figure: (left) The highly mismatched interface material formed by bringing a monolayer of graphene into contact with a monolayer of α-RuCl3. (right) The result (green star) of our ab initio MINT calculation for the graphene/α-RuCl3bilayer added to the Luttinger-Tisza phase diagram from Kim et al. Phys. Rev. B 91, 241110 (2015). The red diamond represents the ground state of plain α-RuCl3. Our MINT calculations indicate that the charge transfer from graphene to α-RuCl3 brings this interface quantum material much closer to the Kitaev point (k) at the origin, relevant to quantum computing applications. |
To overcome this issue, Kim and Arias developed Mismatched Interface Theory (MINT) to study such interfaces theoretically. Our first application of MINT is to the graphene/α-RuCl3 hetero-bilayer interface enabling a quantitative prediction of charge transfer between the two monolayers.
MINT is based on a simple principle and it uses established and widely available standard ab initio methods in each of its steps. Hence MINT is versatile and accessible, and we anticipate the application of this approach to produce many more exciting results in mismatched interface systems previously out of reach of ab initio studies.
E. Gerber et al. Phys. Rev. Lett. 124, 106804 (2020) doi: 10.1103/PhysRevLett.124.106804
Technical Background:
Recent developments in twisted and lattice-mismatched bilayers have revealed a rich phase space of van der Waals systems and generated excitement. Among these systems are heterobilayers, which can offer new opportunities to control van der Waals systems with strong in plane correlations such as spin-orbit-assisted Mott insulator α-RuCl3. Nevertheless, a theoretical ab initio framework for mismatched heterobilayers without even approximate periodicity is sorely lacking. We propose a general strategy for calculating electronic properties of such systems, mismatched interface theory (MINT), and apply it to the graphene=α-RuCl3 (GR=α-RuCl3) heterostructure. Using MINT, we predict uniform doping of 4.77% from graphene to α-RuCl3 and magnetic interactions in α-RuCl3 to shift the system toward the Kitaev point. Hence, we demonstrate that MINT can guide targeted materialization of desired model systems and discuss recent experiments on GR=α-RuCl3 heterostructures.
The work was conducted by the PARADIM in-house research team with additional contributions from Cornell University.
Full Citation:
E. Gerber, Y. Yao, T.A. Arias, and E.-A. Kim, “Ab Initio Mismatched Interface Theory of Graphene on α−RuCl3: Doping and Magnetism,” Phys. Rev. Lett. 124. 106804 (2020)
Return to: In-house Research Highlights



